mutation predictions | marginal predictions | summary statistics | genome diff | command line log
|
breseq version 0.33.1 revision 8505477f25b3
mutation predictions | marginal predictions | summary statistics | genome diff | command line log |
| read file | reads | bases | passed filters | average | longest | mapped | |
|---|---|---|---|---|---|---|---|
| errors | SNFR_19_166_1_S2415_L001_R1_001.good.fq | 999,639 | 140,911,150 | 100.0% | 141.0 bases | 141 bases | 98.8% |
| errors | SNFR_19_166_1_S2415_L001_R2_001.good.fq | 999,639 | 140,911,150 | 100.0% | 141.0 bases | 141 bases | 93.2% |
| total | 1,999,278 | 281,822,300 | 100.0% | 141.0 bases | 141 bases | 96.0% |
| seq id | length | fit mean | fit dispersion | % mapped reads | description | ||
|---|---|---|---|---|---|---|---|
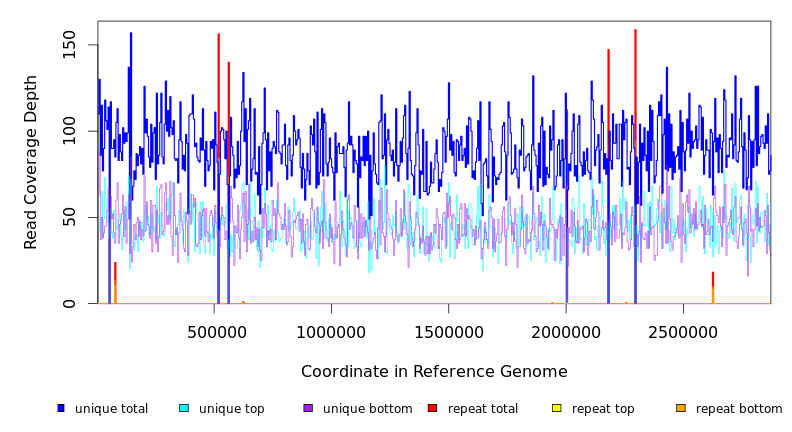
| coverage | distribution | USA300TCH1516_ALE | 2,872,915 | 91.5 | 3.0 | 97.8% | Staphylococcus species strain strain. |
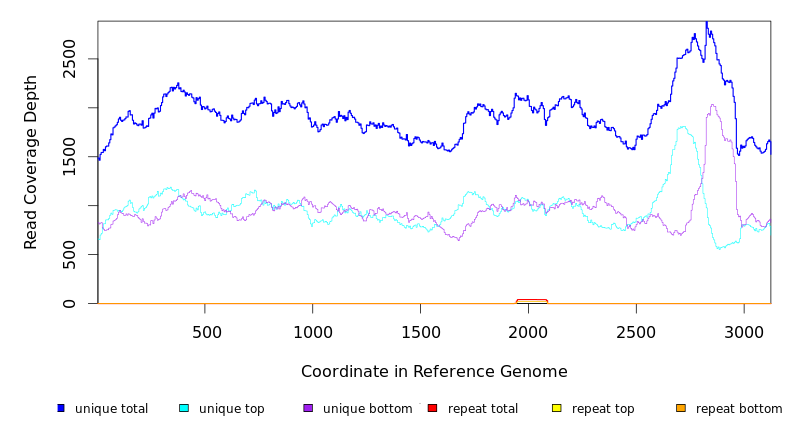
| coverage | distribution | CP000731 | 27,041 | NA | NA | 0.0% | Staphylococcus aureus subsp. aureus USA300_TCH1516 plasmid |
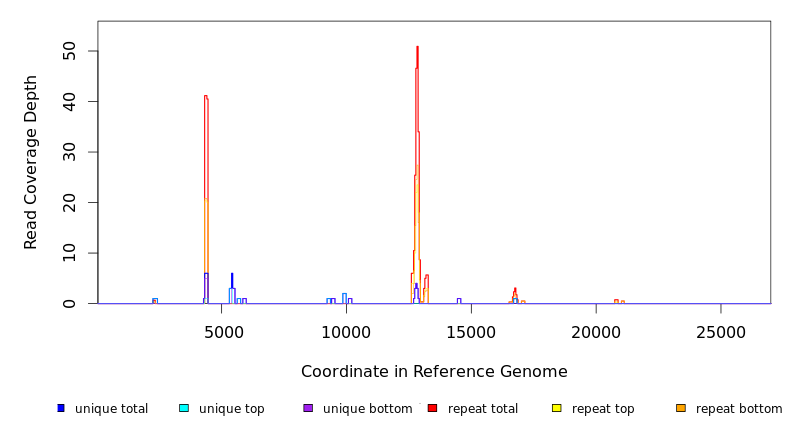
| coverage | distribution | NC_012417 | 3,125 | 1898.9 | 21.9 | 2.2% | Staphylococcus aureus subsp. aureus USA300_TCH1516 plasmid |
| total | 2,903,081 | 100.0% |
fit dispersion is the ratio of the variance to the mean for the negative binomial fit. It is =1 for Poisson and >1 for over-dispersed data.
Insufficient coverage Reference sequence counted as entirely deleted due to low coverage. Try either the -t,--targeted-sequencing or the -c,--contig-reference option if you want mutations called for these reference sequences.
| option | limit | actual |
|---|---|---|
| Number of alignment pairs examined for constructing junction candidates | ≤ 100000 | 8401 |
| Coverage evenness (position-hash) score of junction candidates | ≥ 2 | ≥ 2 |
| Test this many junction candidates (n). May be smaller if not enough passed the coverage evenness threshold | 100 ≤ n ≤ 5000 | 422 |
| Total length of all junction candidates (factor times the reference genome length) | ≤ 0.1 | 0.042 |
| reference sequence | pr(no read start) |
|---|---|
| USA300TCH1516_ALE | 0.81386 |
| CP000731 | NA |
| NC_012417 | 0.11171 |
pr(no read start) is the probability that there will not be an aligned read whose first base matches a given position on a given strand.
| option | value |
|---|---|
| Coverage evenness (position-hash) score of predicted junctions must be | ≥ 3 |
| Skew score of predicted junction (−log10 probability of unusual coverage evenness) must be | ≤ 3 |
| Number of bases that at least one read must overlap each uniquely aligned side of a predicted junction | ≥ 1 |
| option | value |
|---|---|
| Mode | Consensus/Mixed Base |
| Ploidy | 1 (haploid) |
| Consensus mutation E-value cutoff | 10 |
| Consensus frequency cutoff | 0.75 |
| Consensus minimum variant coverage each strand | OFF |
| Consensus minimum total coverage each strand | OFF |
| Consensus minimum variant coverage | OFF |
| Consensus minimum total coverage | OFF |
| Polymorphism E-value cutoff | 10 |
| Polymorphism frequency cutoff | 0.2 |
| Polymorphism minimum variant coverage each strand | OFF |
| Polymorphism minimum total coverage each strand | OFF |
| Polymorphism minimum variant coverage | OFF |
| Polymorphism minimum total coverage | OFF |
| Polymorphism bias cutoff | OFF |
| Predict indel polymorphisms | YES |
| Skip indel polymorphisms in homopolymers runs of | OFF |
| Skip base substitutions when they create a homopolymer flanked on each side by | OFF |
| program | version |
|---|---|
| bowtie2 | 2.3.4.1 |
| R | 3.4.4 |
| step | start | end | elapsed |
|---|---|---|---|
| Read and reference sequence file input | 23:25:19 25 May 2019 | 23:25:50 25 May 2019 | 31 seconds |
| Read alignment to reference genome | 23:25:50 25 May 2019 | 23:30:53 25 May 2019 | 5 minutes 3 seconds |
| Preprocessing alignments for candidate junction identification | 23:30:53 25 May 2019 | 23:31:22 25 May 2019 | 29 seconds |
| Preliminary analysis of coverage distribution | 23:31:22 25 May 2019 | 23:32:29 25 May 2019 | 1 minute 7 seconds |
| Identifying junction candidates | 23:32:29 25 May 2019 | 23:32:39 25 May 2019 | 10 seconds |
| Re-alignment to junction candidates | 23:32:39 25 May 2019 | 23:33:57 25 May 2019 | 1 minute 18 seconds |
| Resolving best read alignments | 23:33:57 25 May 2019 | 23:34:43 25 May 2019 | 46 seconds |
| Creating BAM files | 23:34:43 25 May 2019 | 23:35:40 25 May 2019 | 57 seconds |
| Tabulating error counts | 23:35:40 25 May 2019 | 23:36:01 25 May 2019 | 21 seconds |
| Re-calibrating base error rates | 23:36:01 25 May 2019 | 23:36:03 25 May 2019 | 2 seconds |
| Examining read alignment evidence | 23:36:03 25 May 2019 | 23:40:26 25 May 2019 | 4 minutes 23 seconds |
| Polymorphism statistics | 23:40:26 25 May 2019 | 23:40:26 25 May 2019 | 0 seconds |
| Output | 23:40:26 25 May 2019 | 23:40:34 25 May 2019 | 8 seconds |
| Total | 15 minutes 15 seconds | ||
{kind=link}
{kind=link}
{kind=link}