Sample Resequencing Stats
Note: The mutation counts shown below represent unfiltered mutation sets.
| ALE, Flask, Isolate |
Predicted Mutations |
Mean Coverage |
Total Reads |
Percent Mapped |
Mapped Reads |
Average Read Length |
|
A5 F24 I1 R1
|
10 |
111.5 |
2050540 |
94.5% |
1937760 |
264.9 |
Breseq alignment
BRESEQ :: Evidence
|
| evidence |
seq id |
position |
mutation |
annotation |
gene |
description |
| MC JC |
NC_000913 |
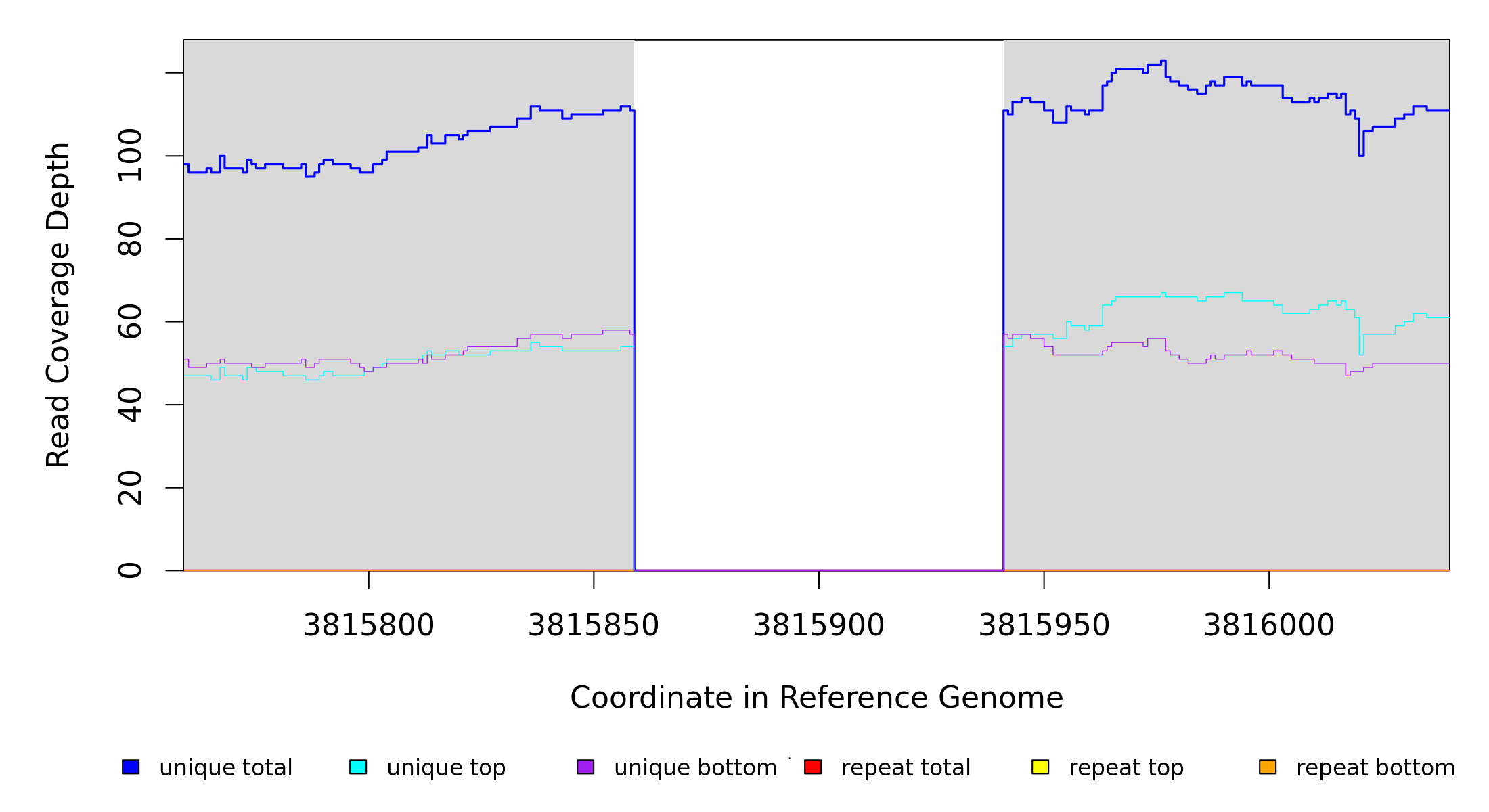
3,815,859 |
Δ82 bp |
|
[rph] |
[rph] |
|
| | | | seq id |
start |
end |
size |
←reads |
reads→ |
gene |
description |
|---|
| * |
* |
÷ |
NC_000913 |
3815859 |
3815940 |
82 |
111 [0] |
[0] 111 |
[rph] |
[rph] |
|
| |
seq id |
position |
reads (cov) |
reads (cov) |
score |
skew |
freq |
annotation |
gene |
product |
| * |
? |
NC_000913 |
= 3815858 | 0 (0.000) | 109 (1.040) |
65/508 |
0.3 |
100% |
intergenic (‑90/+5) |
pyrE/rph |
orotate phosphoribosyltransferase/truncated RNase PH |
| ? | NC_000913 |
3815941 = |
0 (0.000) | coding (609/687 nt) |
rph |
truncated RNase PH |
GATK/CNVnator alignment
BRESEQ :: bam2aln output
GTATTAAACAGCCCGGCGTTGAAGAAATAGGGGCTTTTGCGCCCGGATTTCAGCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGCCGCCTTCTGCGTCGCTACAATGGATTCGATTCCCCTCGGGCCAGAGCCAACAAGATGAGTAGCTCTTCATGGGTGAACGGCTCGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTTCCACCAGCTTCTGTAGCGCATCTACCAGCG > NC_000913/3815644‑3816143
|
GTATTAAACAGCCCGGCGTTGAAGAAATAGGGGCTTTTGCGCCCGGATTTCAGCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCA > M01186:109:000000000‑A8RG1:1:1106:13388:8370/1‑301 (MQ=60)
GTATTAAACAGCCCGGCGTTGAAGAAATAGGGGCTTTTGCGCCCGGATTTCAGCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCA > M01186:109:000000000‑A8RG1:1:1107:21592:10266/1‑301 (MQ=60)
GTATTAAACAGCCCGGCGTTGAAGAAATAGGGGCTTTTGCGCCCGGATTTCAGCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCA > M01186:109:000000000‑A8RG1:1:2114:21783:20031/1‑301 (MQ=60)
TATTAAACAGCCCGGCGTTGAAGAAATAGGGGCTTTTGCGCCCTGATTTCAGCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAG < M01186:109:000000000‑A8RG1:1:1106:13281:5277/301‑1 (MQ=60)
AAATAGGGGCTTTTGCGCCCGGATTTCAGCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTC > M01186:109:000000000‑A8RG1:1:1109:20280:8368/1‑300 (MQ=60)
ATAGGGGCTTTTGCGCCCGGATTTCAGCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACA > M01186:109:000000000‑A8RG1:1:1102:24100:9375/1‑301 (MQ=60)
GCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAG > M01186:109:000000000‑A8RG1:1:2117:20694:4045/1‑250 (MQ=60)
CTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTGCGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGC < M01186:109:000000000‑A8RG1:1:2104:16482:3770/301‑1 (MQ=60)
TGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCT < M01186:109:000000000‑A8RG1:1:2109:22031:12346/301‑1 (MQ=60)
GCTTGCTCAGCGCAAATTCAATAAAGTGGCGCTGATATGGTTTCATGCCTTCGCTCCTGATCGTACTTTTCTACAGACAAAAAAAAGGCCACACATCAGTCGCCTTAAAAATCAGTTTGCCAGAGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCATCTGCCGTCCCCTACACTTCAATGATGCGCCCGTGTTCGGTCATCACTACGTTCCTGTCGGTCTCTGCGGCCGGGTCTTCAACGTATTCCAGATAGAAACCCGCATGGCGGTTCACAATCCCGACAGAAACTGCGGCTACCATCCCTGTCATCGGATTGGTTTTTCGCTT > M01186:109:000000000‑A8RG1:1:2110:5147:17696/1‑301 (MQ=60)
AAGCTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTTCCACCAGCTTCTGT < M01186:109:000000000‑A8RG1:1:2112:16290:9546/301‑1 (MQ=60)
AAACTTGCGATGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTTCCACCAGCTTCTGT < M01186:109:000000000‑A8RG1:1:1114:14894:17160/301‑1 (MQ=60)
ACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTTCCACCAGCTTCTGTAG < M01186:109:000000000‑A8RG1:1:1108:19744:9183/301‑1 (MQ=60)
ACTGGCGCTGAGATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTTCCACCAGCTTCTGTAG < M01186:109:000000000‑A8RG1:1:2107:24179:2683/301‑1 (MQ=60)
GATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCC > M01186:109:000000000‑A8RG1:1:2111:14751:10028/1‑244 (MQ=60)
GATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGAAAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTTCCACCAGCTTCTGTACCGCATCTA > M01186:109:000000000‑A8RG1:1:1105:5682:6840/1‑300 (MQ=60)
GTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTTCCACCAGCTTCTGTAGCCCATCTACCAGCG > M01186:109:000000000‑A8RG1:1:2113:14529:3844/1‑300 (MQ=60)
GTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGC‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑CGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTCCCACCACTTTCTGTCGCGCACCTACCAGCG > M01186:109:000000000‑A8RG1:1:1112:20023:24266/1‑300 (MQ=60)
|
GTATTAAACAGCCCGGCGTTGAAGAAATAGGGGCTTTTGCGCCCGGATTTCAGCGTAAACTCGCCAAACTTTAACACCTGCTTGCTAAGCGCAAATTCAATAAACTGGCGCTGATATGGTTTCATGCCTTCGCTCCTCATCTTACTTTTCTACAGACAAAAAAAAGGCGACTCATCAGTCGCCTTAAAAATCAGTTTGCCAGCGCCGCCTTCTGCGTCGCTACAATGGATTCGATTCCCCTCGGGCCAGAGCCAACAAGATGAGTAGCTCTTCATGGGTGAACGGCTCGCCTTCTGCCGTCCCCTGCACTTCAATGATGCGCCCGTCTTCGGTCATCACTACGTTCATGTCGGTCTCTGCGGCAGAGTCTTCAACGTATTCCAGATCGCAAACCGCTTCGCCGTTCACAATTCCGACAGAAACTGCGGCTACCATCCCTTTCATCGGATTGGTTTTCAGCTTGCCGTTTTCCACCAGCTTCTGTAGCGCATCTACCAGCG > NC_000913/3815644‑3816143
|
| Alignment Legend |
|---|
Aligned base mismatch/match (shaded by quality score): ATCG/ATCG < 0 ≤ ATCG/ATCG < 9 ≤ ATCG/ATCG < 21 ≤ ATCG/ATCG < 35 ≤ ATCG/ATCG < 38 ≤ ATCG/ATCG |
Unaligned base: atcg Masked matching base: atcg Alignment gap: ‑ Deleted base: ‑ |