Sample Resequencing Stats
Note: The mutation counts shown below represent unfiltered mutation sets.
| ALE, Flask, Isolate |
Predicted Mutations |
Mean Coverage |
Total Reads |
Percent Mapped |
Mapped Reads |
Average Read Length |
|
A1 F1 I179 R2
|
4 |
15.4 |
992998 |
94.0% |
933418 |
83.4 |
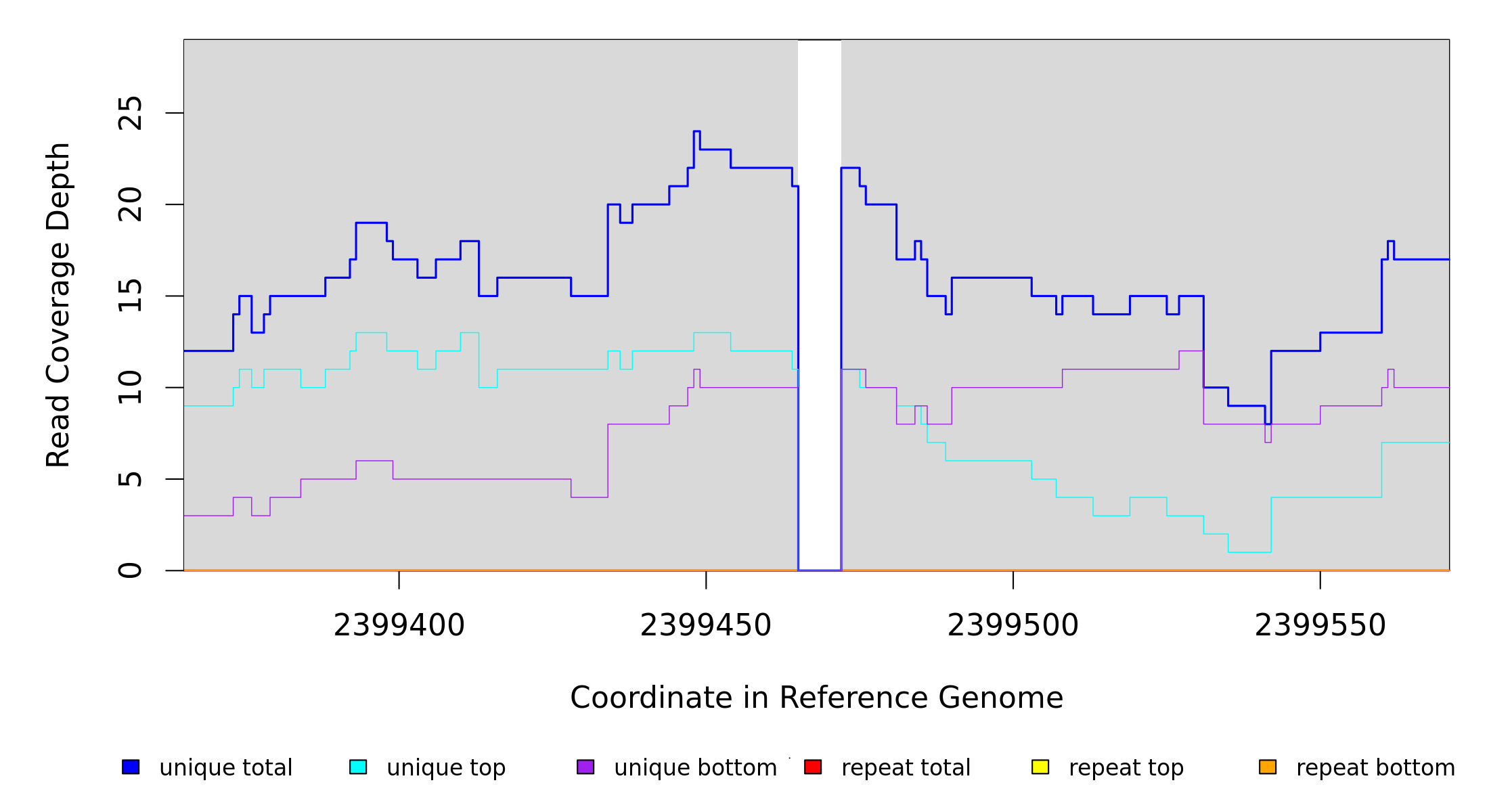
Breseq alignment
BRESEQ :: Evidence
|
| evidence |
seq id |
position |
mutation |
annotation |
gene |
description |
| MC JC |
NZ_CP009273 |
2,399,465 |
Δ7 bp |
coding (650‑656/939 nt) |
lrhA ← |
transcriptional regulator LrhA |
|
| | | | seq id |
start |
end |
size |
←reads |
reads→ |
gene |
description |
|---|
| * |
* |
÷ |
NZ_CP009273 |
2399465 |
2399471 |
7 |
21 [0] |
[0] 22 |
lrhA |
transcriptional regulator LrhA |
|
| |
seq id |
position |
reads (cov) |
reads (cov) |
score |
skew |
freq |
annotation |
gene |
product |
| * |
? |
NZ_CP009273 |
= 2399464 | 0 (0.000) | 21 (1.280) |
17/164 |
0.1 |
100% |
coding (657/939 nt) |
lrhA |
transcriptional regulator LrhA |
| ? | NZ_CP009273 |
2399472 = |
0 (0.000) | coding (649/939 nt) |
lrhA |
transcriptional regulator LrhA |
GATK/CNVnator alignment
BRESEQ :: bam2aln output
CGCAGGTCCGGGCTCATCATCTCAACCGGCCTTGCCGTCACGCCAAGACCGGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAGGCGACATAAGCCAGTCGCCATGGAATATCTGCTTTATTAAGCGTCGCCAGCACCATATCGCGAAACGGGCTAGGAT > NZ_CP009273/2399378‑2399540
|
CGCAGGTCCGGGCTCATCATCTCAACCGGCCTTGCCGTCACGCCAAGACCGGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCA > SRR3722052.395233/1‑100 (MQ=60)
GCAGGTCCGGGCTCATCATCTCAACCGGCCTTGCCGTCACGCCAAGACCGGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCAT < SRR3722052.356178/100‑1 (MQ=60)
GGTCCGGGCTCATCATCTCAACCGGCCTTGCCGTCACGCCAAGACCGGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCATGGA > SRR3722052.64847/1‑100 (MQ=60)
GTCCGGGCTCATCATCTCAACCGGCCTTGCCGTCACGCCAAGACCGGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCATGGAA > SRR3722052.219011/1‑100 (MQ=60)
ATCTCAACCGGCCTTGCCGTCACGCCAAGACCGGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCATGGAATATCTGCTTTATT > SRR3722052.177860/1‑100 (MQ=60)
GACCGGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCATGGAATATCTGCTgtctcttatacacatctccgagcccacgagaca > SRR3722052.213231/1‑67 (MQ=60)
GGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCATGGAATATCTGCTTTATTAAGCGTCGCCAGCACCATATCGCGAAACGGGC > SRR3722052.409765/1‑100 (MQ=60)
acagGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCATGGAATATCTGCTTTATTAAGCGTCGCCAGCACCATATCGCGAAACGGGCTAGGAT < SRR3722052.105690/96‑1 (MQ=60)
CACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCATGGAATATCTGCTTTATTAAGCGTCGCCAGCACCATATCGCGAAACGGGCTAGGAT < SRR3722052.151775/100‑1 (MQ=60)
CACTGCCGCACGAACGGCCGGAAGCGTCGAG‑‑‑‑‑‑‑AAGCCAGTCGCCATGGAATATCTGCTTTATTAAGCGTCGCCAGCACCATATCGCGAAACGGGCTAGGAT < SRR3722052.1873/100‑1 (MQ=60)
|
CGCAGGTCCGGGCTCATCATCTCAACCGGCCTTGCCGTCACGCCAAGACCGGCTTTCACTGCCGCACGAACGGCCGGAAGCGTCGAGGCGACATAAGCCAGTCGCCATGGAATATCTGCTTTATTAAGCGTCGCCAGCACCATATCGCGAAACGGGCTAGGAT > NZ_CP009273/2399378‑2399540
|
| Alignment Legend |
|---|
Aligned base mismatch/match (shaded by quality score): ATCG/ATCG < 0 ≤ ATCG/ATCG < 33 ≤ ATCG/ATCG < 34 ≤ ATCG/ATCG < 35 ≤ ATCG/ATCG < 41 ≤ ATCG/ATCG |
Unaligned base: atcg Masked matching base: atcg Alignment gap: ‑ Deleted base: ‑ |